I used to teach sections 1 to 4 to Year 2 student engineers as preparation for lectures on psychrometry (air conditioning) and hydrocarbon chemistry. For a while, subject to available lecture slots, I introduced the Second Law topic of Gibbs function as an aside. Successive COPs and the urgency of climate change will inevitably reduce the need for engineers who design fossil-fuel fired power stations. The UK, I believe, will need engineers who can carry out lifecycle analysis of existing stock, and inform international diplomacy and negotiation. With this in mind some preliminary knowlege of Gibbs function, chemical equilibrium, and the indicative quantities of pollutants produced is more useful than used to be the case. The Second Law analysis of chemical reactions has always been a feature of electrochemistry - with the world's increased reliance on batteries, fuel cells and electrolysers it has become an important part of the General Engineering syllabus.
2. Introduction
Gas mixtures are found when hydrocarbons are used in internal combustion engines, in air conditioning where "moist air" is envisaged as a mixture of steam and "dry air", and in fuel cells. To carry out energy balances, engineers need to calculate the enthalpy content of gas mixtures. Such is our principal aim here, we also touch on Second Law topics frequently found in published literature. (1) When gases are mixed there is a change in entropy. (2) When gases react chemically they have a potential to transfer heat (the enthalpy change). (3) During chemical reaction, gases have a potential to fuel useful work. The appropriate, new, thermodynamc property is termed the "Gibbs Free Energy" or the "Gibbs function".
Students should be able to:
work interchangeably between properties defined in terms of mass (kg) and properties defined in terms of substance (kmol);
describe the assumptions and consequences of the Gibbs-Dalton law;
describe and calculate partial pressure, mass fraction and mole fraction;
for an ideal mixture of gases, calculate specific properties (i.e. "per kg" properties),\(c_v, c_p, R, h, u \).
make a "guestimate" of the impact of chemical dissociation.
Engineers need to work interchangeably with mass (kg) and substance (kmol)
The basic chemistry taught in high school should be well understood . The gram mole is the SI unit of substance, equivalent to \( 6.022 \times 10^{23}\) atoms or molecules. Thus the atomic mass of carbon is the mass of \(6.022 \times 10^{23}\) carbon atoms, which is 12 grams. Similarly the atomic mass of hydrogen is the mass of the same number of hydrogen atoms: 1 gram approx.
Relative molecular mass is the mass of a single molecule divided by (1/12) times the mass of a single carbon atom. It has no dimensions. Molar mass, on the other hand, has units of g per mol or kg per kmol. Relative molecular mass is found by summing the atomic masses of constituent elements.
Sample Calculations of Relative Molecular Mass
Substance
Structure
Relative Atomic Mass
Elemental Hydrogen
H.
1.008
Elemental Carbon
C
12
Elemental Nitrogen
N.
14
Elemental Oxygen
O.
16
-
-
Relative Molecular Mass
Oxygen
\(O_2 \qquad O=O\)
\( 2 \times 16=32\)
Nitrogen
\(N_2 \qquad N \equiv N \)
\( 2 \times 14=28\)
Methane
\(CH_4\)
\( 12 +4\times 1.008 \approx 16\)
Octane
\(C_8H_{18}\)
\( 8\times12 +18\times 1.008 \approx 114\)
Carbon Dioxide
\( CO_2 \qquad O=C=O \)
\( 12 + 2 \times 16 = 44 \)
The amount of substance of species i is equal to its mass divided by its molar mass,
$$ n_i = \frac{m_i}{\widetilde{m}_i} $$
For example, 64 kg of methane are equivalent to 64/16=4 kmol.
In a mixture of two or more species the mole fraction of species i is defined as:
$$ mole \; fraction \; of \; i \; = \frac{\; moles \; of \; i}{ moles \; of \; all \; species }$$
Or
\begin{equation*}
\widetilde{x}_i=\frac{n_i}{n}
\end{equation*}
One also works with the mass fraction of species i:
$$ mass \; fraction \; of \; i \; = \frac{\; mass \; of \; i}{ mass \; of \; all \; species }$$
Or
\begin{equation*}
x_i=\frac{m_i}{m}
\end{equation*}
Example 4B.010: Mole fractions. A mixture contains 36 kg of water vapour and 66 kg of carbon dioxide. Find the mass fractions of each. Find the mole fractions of each. What is the relative molecular mass of the mixture?)
Solution: The molar mass of water is \(2\times 1+16 = 18 kg/kmol\) and the molar mass of carbon dioxide is \(12 + 2\times 16 = 44 kg/ kmol\). In our mixture:
Amount of water (in kmol)
\begin{align*}
n_{H_2O}=& \frac{36kg(H_2O)}{18 kg(H_2O)/kmol(H_2O)} \\
=&2 kmol (H_2O)
\end{align*}
Amount of CO2 (in kmol)
\begin{align*}
n_{CO_2}=&\frac{66 kg(CO_2)}{44 kg(CO_2)/kmol(CO_2)} \\
=&1.5 kmol (CO_2)
\end{align*}
Mole fractions of water and carbon dioxide
\begin{align*}
\widetilde{x}_{H2O} = \frac{n_{H2O}}{n} =2/(2+1.5)=0.571 \\
\widetilde{x}_{CO2} = \frac{n_{CO2}}{n} =1.5/(2+1.5) =0.429
\end{align*}
The mass of mixture is 36 + 66 = 102 kg and the amount of substance is 2 + 1.5 = 3.5 kmol. The molar mass (mass per kmol) is
\begin{equation*}
\widetilde{m} = \frac{102kg}{3.5kmol}=29.14 kg/mol
\end{equation*}
4. The Gibbs-Dalton Law
'The pressure and internal energy of a mixture of gases are respectively equal to the pressures and internal energies of the individual constituents when each occupies a volume equal to that of the mixture at the temperature of the mixture.' ( Rogers and Mayhew )
Gibbs-Dalton usefully helps us estimate the properties of ideal gas mixtures when given the properties of the pure components. Such properties are part of the
'Steam Tables' published by Rogers and Mayhew.
Consider a volume, containing \(n_i\) moles of chemical species i plus a quantity of other species, and at total pressure p. If only i were present, and i expanded to fill the same volume and at the same temperature, the pressure would be reduced to \(p_i\); term \(p_i\) is called the 'partial pressure'. (Note that the expansion is irreversible). Similarly the pure gas would have an internal energy \(U_i\). As formulae, the Gibbs Dalton law can be written as:
\begin{align}
U =& \sum U_i]_{V,T} \\
p =& \sum p_i]_{V,T} \qquad (1)
\end{align}
Figure 1. Visual representation of the Gibbs-Dalton Law. Vessel "mix" holds a mixture of gases 1 and 2. Vessels 1 and 2
hold equivalent masses of pure 1 and pure 2 respectively, at the same temperature and volume. \( \qquad p_{mix}=p_1+p_2 \qquad U_{mix}=U_1+U_2\)
Note that in equation 1 "i]V,T" means "as if species i were, by itself, to fill volume V at temperature T".
Gibbs-Dalton shows (1) that a specific property of a mixture (\(c_v , c_p, u, R, h\)) is a weighted mean of the specific property of the constituents (entropy forms the exception) (2) that partial pressure is the product of total pressure and mole fraction (3) that partial pressure of a constituent follows a similar form to the Ideal Gas Law ( or equation of state).
The Gibbs-Dalton Law and Partial Pressure
For a component of a gas mixture, i, consider an equal mass of pure component i held in a system of identical volume and temperature. The pressure therein is termed the "partial pressure of i", \(p_i\).
Start with Ideal Gas Law for a mixture of perfect gases, where terms m, R, V are respectively the mass, specific gas constant and volume of the mixture. Mechanical Engineers usually use the ideal gas law in the form \( pV = mRT \) but here it is more convenient to use molar properties,
\begin{equation}
p = n \frac{\widetilde{R}T}{V} \qquad (2)
\end{equation}
According to the definition of partial pressure, and assuming ideal gases with no molecule-to-molecule forces, then a component (or species) i holds a partial pressure according to
\begin{equation}
p_i = n_i \frac{\widetilde{R}T}{V} \qquad (3)
\end{equation}
In other words, \(p_i\) is the pressure that component i would exert were it acting as the only (pure) component within volume V (at temperature T). Let us divide the molar form of Equation 3 throughout by the molar form of Equation 2.
\begin{equation}
\frac{p_i}{p}=\frac{n_i}{n}=\widetilde{x}_i \qquad (3B)
\end{equation}
The final term is the mole fraction, \(\widetilde{x}_i\).
The Gibbs-Dalton Law and Properties
Most specific ("per kg") properties of a mixture are an average of the corresponding
pure component properties, weighted by mass fractions.
Gibbs Dalton gives us the properties of gas mixtures. The properties are weighted by mass fraction. For example, consider the specific gas constant, R. One contrasts the mixture gas constant with the individual species values. Here, let us return
to the "General Engineers form" of the IGL, \( pV = mRT \), for both the mixture and individual species. Given that the total pressure is the sum of individual partial pressures,
\begin {equation*}
p = \frac{T}{V}mR = \sum p_i = \frac{T}{V} \sum m_i \, R_i
\end{equation*}
(The \(]_{V,T}\) qualification is omitted from pressure.) The common factor of (T/V) enables rearrangement to give,
where \(x_i\) is the mass fraction of component i (= mass of component i / total mixture mass). A similar approach applies to the specific heat capacity at constant volume.
\begin{equation}
U = m \, c_v \, T = \sum U_i = \sum m_i \, c_{v,i} \, T
\end{equation}
(The \(]_{V,T}\) qualification is omitted from internal energy.) On rearrangement,
This form applies to most specific properties (where units include \( kg^{-1}\)) - \( v, c_p, h, u \) (but not specific entropy, s).
Amgat’s Law of Partial Volumes
A similar concept to partial pressure is partial volume, \(V_i\). This time consider n moles of a mixture at pressure p, volume V and Temperature T. If all species except i are removed, and p,T are unchanged, the volume is reduced to \(V_i\). Amgat's law gives.
Volumetric analysis is often quoted in the field of combustion.
Example 4B.020: Partial pressures, partial volumes and enthalpy of a gas mixture. Consider a mixture of 4 kg methane, 11 kg carbon dioxide, 3.6 kg water vapour, temperature T = 500 K, and volume V = 3 cubic metres. Find partial pressures and volumes. Find change in enthalpy when temperature is raised from 500 to 600K. Find the specific heat capacity of the gas mixture.
Solution:
Molar masses (in kg/kmol) are: methane, \(12 + 4 \times 1=16\); carbon dioxide, \(12 + 2 \times 16 =44\); water, \(16 + 2 \times 1 = 18\). The amount of substance is ...
methane, \(n_{CH4} = 4/16 = 0.25 kmol\); carbon dioxide, \(n_{CO2} = 11/ 44 = 0.25 kmol\); water vapour, \(n_{H2O} = 3.6/18 = 0.2 kmol\). Total amount of substance, n = 0.25 + 0.25 + 0.20 = 0.7 kmol.
The mole fraction of methane is \( \widetilde{x}_{CH4}=0.25/0.7=0.357\). Similarly the mole fractions of carbon dioxide and water are 0.357 and 0.2/0.7 = 0.286 respectively.
Use the Ideal Gas Law to get the total pressure, then use mole fractions to get partial pressures.
\begin{align*}
p =& n \widetilde{R} T/V = 0.7 kmol \times 8.314 kJ kmol^{-1} K^{-1} \times 500 K / 3 m^3 \\
=& 967 kN m^{-2} = 9.67 bar \\ \\
p_{CH4}=&\widetilde{x}_{CH4} \times p = 0.357 \times 9.67 = 3.46 bar \\
p_{CO2} =&\widetilde{x}_{CO2} \times p = 0.357 \times 9.67 = 3.46 bar \\
p_{H2O} =&\widetilde{x}_{H2O} \times p = 0.286 \times 9.67 = 2.77 bar
\end{align*}
To obtain the enthalpy change I uses the specific heat capacities given on page 17 of a set of
'Steam Tables' .
At 550 K they are 3.074, 1.046 and 1.984 kJ/Kg K for methane, carbon dioxide and water vapour.
The mixing of two different types of fluid produces an increase in total entropy.
An understanding of the entropy of mixing helps the reader to tackle certain articles in journal papers.
Firstly, depending on the degree of dilution energy is required to purify gases (e.g. to remove carbon dioxide or sulphur dioxide from air). The entropy of mixing hints that gas purification is best tackled with more concentrated mixtures of gases.
Secondly, the quoted exergetic values of fuels allow for the entropy of mixing.
Thirdly, entropy is an important aspect of chemical equilibrium.
Entropy of pure gases
A fluid's isothermal entropy change is the quotient of reversible work and temperature.
A
previous description of entropy is "reframed" to emphasise isothermal work and molar quantities. The
formal definition of entropy is,
where \(S_o^ø\) is entropy at a standard state" which by convention is standard temperature \(T \equiv T_o = 298K \) and standard pressure \(p \equiv p^ø = 1 bar \).
Consider a constant volume process concerning n kmol of substance. For a small amount of heat transfer \(dQ = n \widetilde{c}_v dT \) so that
$$ S-S_o = n \widetilde{c}_v \int_{o} \frac {dT}{T} = n \widetilde{c}_v ln (\frac{T}{T_o}) \qquad constant \; volume \qquad (8) $$
is the constant volume entropy change from standard temperature \(T_o\) to temperature T, and
where
\( \widetilde{c}_v dT \)
is the isochoric molar heat capacity.
For an isobaric process we use the isobaric molar heat capacity.
$$ S-S_o^p = n \widetilde{c}_p \int_o \frac {dT}{T} = n \widetilde{c}_p ln (\frac{T}{T_o}) \qquad constant \; pressure \qquad (9) $$
is the entropy change at constant pressure, p, from standard temperature \(T_o\) to temperature T.
In chemical equilibrium we shall consider entropy changes under isothermal conditions. For moving boundary work relating to closed systems integration of the area under the pV curve
yields,
$$ W_{rev} = -\int_ø p dV = -n\widetilde{R} \int_ø \frac{dV}{V} =-n \widetilde{R} ln( \frac{V}{V^ø} )= +n \widetilde{R}T ln \frac{p}{p^ø} \qquad constant \; temperature $$
where the result is the net isothermal reversible work of compression. In short,
$$ W_{rev} = n \widetilde{R}T ln \frac{p}{p^ø} \qquad constant \; temperature \; (10) $$
It will be useful to note that the result for the shaft work in open systems is identical,
$$ W_{rev,shaft} = +\int_ø V dp = +n\widetilde{R} T \int_ø \frac{dp}{p} = n \widetilde{R}T ln \frac{p}{p^ø} $$
Under isothermal conditions, the reversible work is equal to the reversible heat addition (times -1). The constant temperature T facilitates integration,
Example: Estimate the molar entropy of oxygen at (1) 400 K and a pressure of 1 bar (2) 298 K and a pressure of 5 bar. (Use
\( \widetilde{s}^ø_o=205.03kJ/kmol K \; and \; \widetilde{c}_p =29.744 kJ/kmol K \)
).
The constant volume mixing of two quantities of the same type of liquid at different temperatures provides a simple illustration.
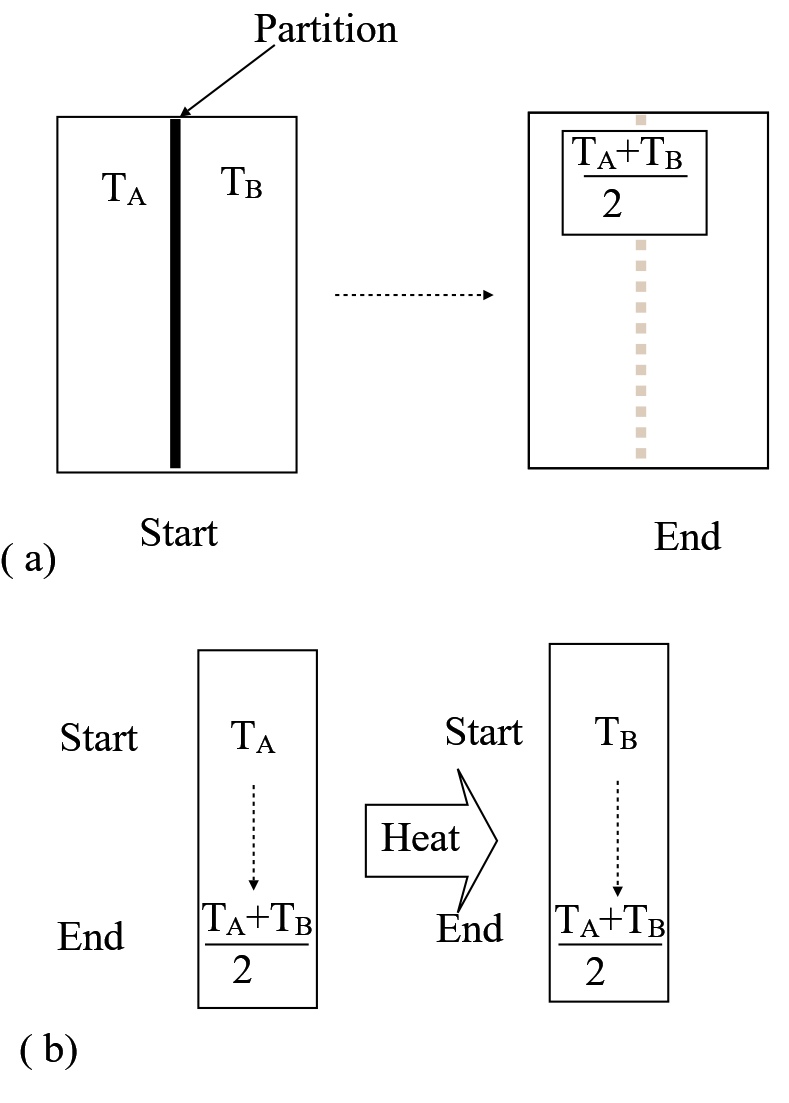
Consider two different processes that achieve the same change in state. In Figure 2 a partitioned vessel holds two equal amounts of liquid at separate temperatures, \(T_A,T_B\). Removing the partition allows the two amounts to mix and achieve the average temperature, \( (T_A+T_B)/2 \). The same result can be brought about if A and B are held in physically separate vessels (Figure 2b). Net heat transfer from A to B ultimately
brings both masses to the same intermediate temperature as achieved by mixing, \(T_A/2+T_B/2\). Therefore the same change in total entropy applies to both heat transfer and mixing. Equation 8 (above) yields,
$$ \Delta S_{tot} = n \widetilde{c}_v
( ln(\frac{T_A+T_B}{2T_A}) + ln(\frac{T_A+T_B}{2T_B}) = n \widetilde{c}_v ln ( \frac{(T_A+T_B)^2}{4 T_A T_B} ) $$
where n is the amount of either one of the two fluids. So long as \( T_A \neq T_B\) the total entropy change exceeds zero, meaning that both Figure 2a and Figure 2b show irreversible processes. The irreversibility can be attributed to mixing in part 2a and heat transfer in part 2b.
Figure 2. Mixing of two equal masses of fluid at different temperatures (a) irreversible heat transfer between separate masses at \(T_A \) and \( T_B\) (b) equivalent irreversible outcome by removing a partition between seperate masses at temperatures \(T_A\) and \(T_B\)
Mixing of different chemical species
... is thermodynamically irreversible.
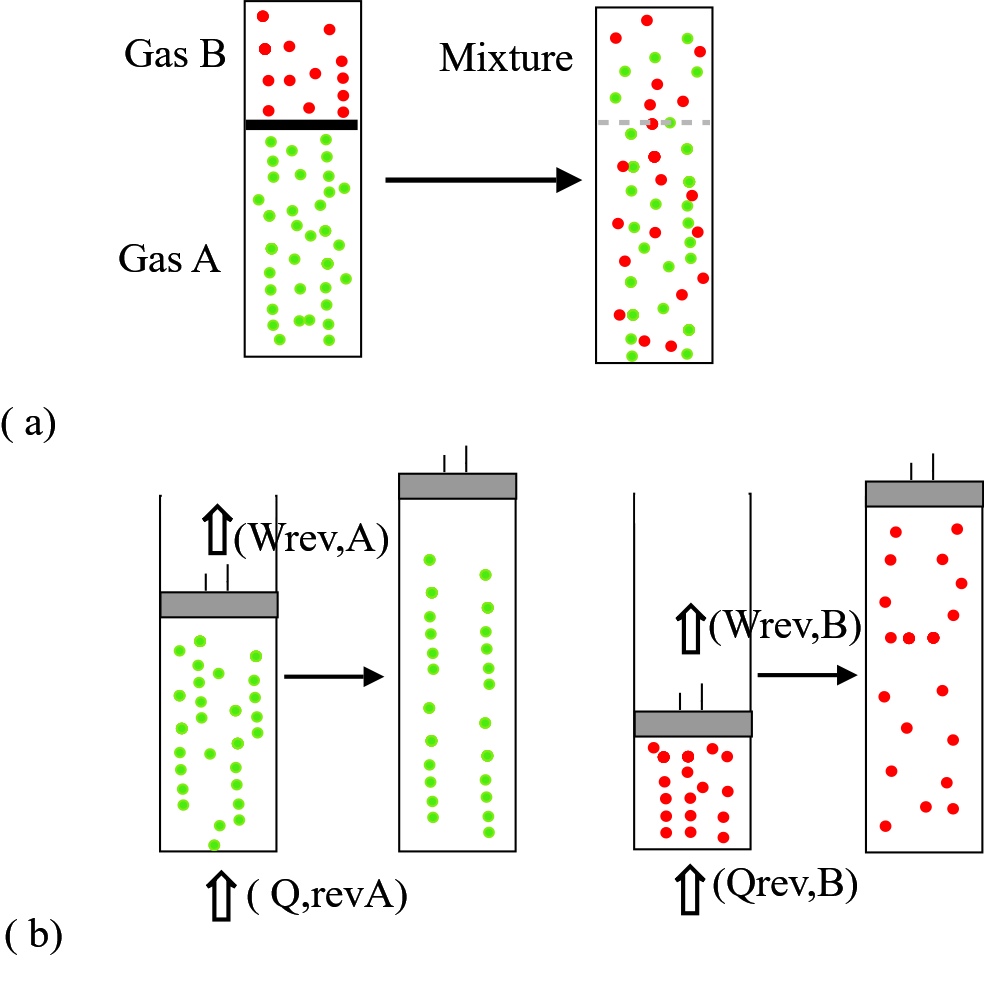
Having shown that mixing fluids at different temperature generates entropy, let us now do the same for fluids of different chemical form. Consider a rigid container holding ideal gases A and B, the two separated by a partition and with each gas at pressure p (Figure 3a). Removal of the partition would allow A and B to expand and occupy the entire vessel with respective partial pressures \(p_A \; and \; p_B \). The process is known to be practically adiabatic and irreversible.
The entropy change, however, can be estimated by considering equivalent reversible expansions of isolated pure gases shown in Figure 3b.
From Equation 10 the total isothermal reversible work for two gases is
$$ W_{rev} = \widetilde{R} T (n_A ln (p_A/p) + n_B ln (p_B/p)) $$
The above ratios of pressure are equal to mole fraction (Equation 3B ),
$$ W_{rev} = \widetilde{R} T (n_A ln (\widetilde{x}_A) + n_B ln (\widetilde{x}_B)) $$
Then (given that the logarithms of mole fractions are negative) \( \Delta S > 0 \). Increased entropy indicates a thermodynamically irreversible process.
Figure 3. Isothermal mixing of different gases (a) reversible equivalent processes whereby A and B are expanded to fill the complete volume (b) equivalent irreversible outcome by removing a partition between A and B.
Example 4B.030: Find the minimum, reversible work required to separate at Standard Ambient Temperature and Pressure (SATP) (a) 1kmol of carbon dioxide from 2500 kmol air (b) 1kmol of carbon dioxide from 8.7 kmol of exhaust gases.
Solution: This concerns compression of gas rather than the expansion associated with mixing. Other than CO2, treat the components of air or exhaust gas as indistinguishable. In other words, assume that this
particular part of the mixture behaves as a pure gas. The minimum work required
is the reversible work of mixing multiplied by (-1).
$$ W_{rev,comp} = (-1) \times \widetilde{R} T (n_A ln (\widetilde{x}_A) + n_B ln (\widetilde{x}_A) ) $$
Where A represents CO2 and B other gases. For the most dilute case, case (a)
The above excludes the massive irreversibilities in separation processes, and the further work needed to compress the CO2 for storage. The comparison does offer a very broad hint that may be more easily capturedirectly from power stations from ambient air by photosynthesis or
direct air capture .
Be aware of the "Gibbs paradox". Suppose our two gases are not distinct, for example both gases A and B are hydgrogen. We then estimate a change in entropy that clearly does not exist. This can be discussed rigorously only by recourse to
statistical thermodynamicists.
6. Gas Mixtures and Chemical Reactions.
Energies of Chemical Reaction (Enthalpy and Gibbs Function)
The maximum heat transfer from a chemical reaction is the enthalpy of reaction. The theoretical maximum, reversible work from an equivalent reaction is the Gibbs function of reaction. Gibbs function is a potential to do work.[
'Statement qualifications apply' .]
For the same start and end (T,p), a thought experiment demonstrates that (1) the products of an exothermic (heat yielding) chemical reaction have lower entropy than their reactants (2) there is a reduction in enthalpy termed the "enthalpy of reaction" (3) the maximum, reversible work achievable is less than the heat of reaction (4) there is a further property, Gibbs Function, that is the potential of a gas mixture to do reversible work (Gibbs function is also the "driving force" for a chemical reaction.)
Consider a hydrogen fueled heat engine. If this operates reversibly and on a set of reversible heat engines, then the conversion of heat to work is no different to that from a theoretically reversible electrochemical device (namely a fuel cell). The chemical equation is simple.
H2 + ½ O2 → H2O
1 kmol H2 + ½ kmol O2 produces 1 kmol steam
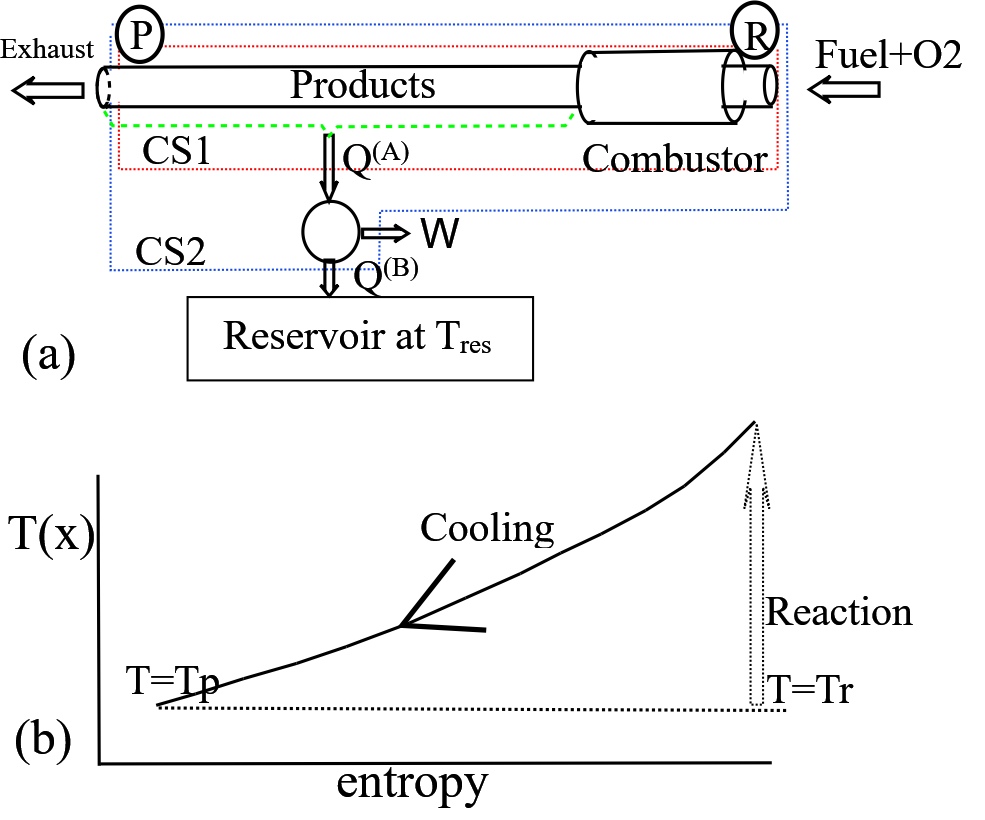
On Figure 4, let 1 kmol hydrogen and 1/2 kmol oxygen enter a pipe at temperature \(T_R\) and position R. They react reversibly and chemically, and as they pass through a tube the chemical species reject a net amount of heat, \(Q_A\) until achieving temperature \(T_P\) and position P and all reactants converted to steam. (Figure 4 is simplified - a more complicated arrangement would be needed to guarantee that the chemical reaction itself is thermodynamcally reversible.) The rejected heat, \(Q^{(A)} \), is supplied to a reversible engine - for example an infinite set of Carnot engines - to obtain theoretical maximum work. The
Kelvin Plank statement mandates further net heat rejection as \( Q ^{(B)}\)to a reservoir at temperature \(T_{res}\).
Figure 4 Demonstrating energy balances on reactants (H2 and O2) and product (H2O) at the same temperature. (a) Hypothetical plant - reactant enters from the right at temperature \(T_R\) and is eventually converted to steam at temperature \(T_P\). All heat produced, \( Q ^{(A)}\), drives a reversible engine which in turn rejects heat to a reservoir at temperature \(T_{res}\).
Take energy balances (or SFEE) for the two boundaries, CS1 and CS2.
$$ Q ^{(A)} = H_P - H_R \qquad heat \; of \; reaction\; CS1 $$
$$ W_{rev} = H_P - H_R - Q ^{(B)} \qquad reversibe \; work \; of \; reaction \;CS2$$
Term \( Q ^{(A)} \) is related to enthalpy - a property - and is termed the "heat" or "enthalpy" of reaction when \(T_P= T_R\). To relate \( W_{rev} \) to properties consider the change in entropy between the fuel and the reactants. Let us assume that the reservoir, reactants and final products all share the same temperature,
( To check consistency, note for reversible processes that
the Eight Corrolary of the Second Law
implies zero total entropy production and hence \( T\Delta S_{res} = -1 \times (S_P-S_R) = -Q^{(B)} \). )
Combine the preceeding two expressions for \( Q ^{(B)}, W_{rev} \) ,
$$ W_{rev} = H_P-H_R - T (S_P-S_R) = [H_P - T S_P] - [H_R - T S_R] = G_P - G_R $$
The terms in brackets[] are termed Gibbs Free Energy or Gibbs Function . They represent the potential of a material to cause isothermal, isobaric, reversible work. More generally, for any temperature T,
$$ G = H - T S $$
Methods of Calculation
The necessary and frequently tabulated data are heat of reaction at standard state, molar enthalpy as a function of temperature, and molar entropy at standard pressure as a function of temperature. [
'Statement qualifications apply' .]
The enthalpies of reaction are often tabulated for "standard" conditions. These are \(T_o=298 K\) and \(p^{\text{ø}}=1bar\). For a basis of \(n_{fuel} = 1 kmol \; of \; fuel \),
where subscript P0 refers to the enthalpies and Gibbs functions of products at the standard temperature (298 K), superscript ø specifies standard pressure (1 bar) and R0 refers to reactants. For ideal gases enthalpy is independent of pressure and
the superscript ø can be omitted. The term
\( \Delta \widetilde{h}_o^{\text{ø}} \) is the standard enthalpy of reaction (or combustion), and \( \widetilde{s}_{o,i}^{\text{ø}} \)
is the standard molar entropy of species i at standard state; check that any tabulated molar entropy is zero at 0K and 1 bar. Also
\( \Delta n_i \) is the net amount of species i created by the reaction of 1kmol of fuel; it has positive sign for products and negative sign for reactants. For pure ideal gases enthalpy depends on temperature only and no correction for pressure is needed. You will very rarely need to
correct the Gibbs function for a change in reference pressure - remember that the reference pressure refers simply to tabulated and database values.
For example, consider the chemical conversion of hydrogen and oxygen to water in a fuel cell. Approximately, pressure = 1 bar is constant and reactants and products enter and leave at the same temperature, 298.15K. Rogers and Mayhew (page 361) tabulate a limited set of enthalpies and Gibbs function, and at the "standard state" conditions here selected the enthalpies of the elements (in their natural state) are zero.
H2 + ½ O2 → H2O
-1 kmol -½ kmol +1 kmol
\( T \widetilde{s}_{o,h2}^{\text{ø}}=38931\) \( T \widetilde{s}_{ o,o2}^{\text{ø}}=61131 \) \( T\widetilde{s}_{ o,h2o}^{\text{ø}}=56268\) kJ/kmol
So for a perfect fuel cell the approximate available heating effect is \( H_P-H_R \approx -242-0-0/2 \approx -242 MJ/kmol(fuel) \) but the theoretical available electrical power is slightly less at \( G_P-G_R \approx -242 - (56-39-61/2) = -228MJ/kmol(fuel) \). The theoretical efficiency of the fuel cell is 95%. (Following some conventions, we have ficticiously treated the moisture as if it remains in the vapour phase. The quoted enthalpy of combustion is related to the "lower heating value" (US) or "net calorific value" (UK). The "higher heating value" or "gross calorific value" would allow for the heat released by moisture condensation".) The real world efficiency is closer to 60%.
So what happens if the reaction temperature is hotter or colder than 298K? It is often the case that sensible enthalpies are reported over a range of temperatures (or molar heat capacities are
reported) but enthalpies of reaction are reported only for standard conditions. We can correct the enthalpy of reaction to account for the sensible enthalpies of the reactants and products. The sensible enthalpies should have a temperature datum at 298K (pressure has no effect for ideal gases).
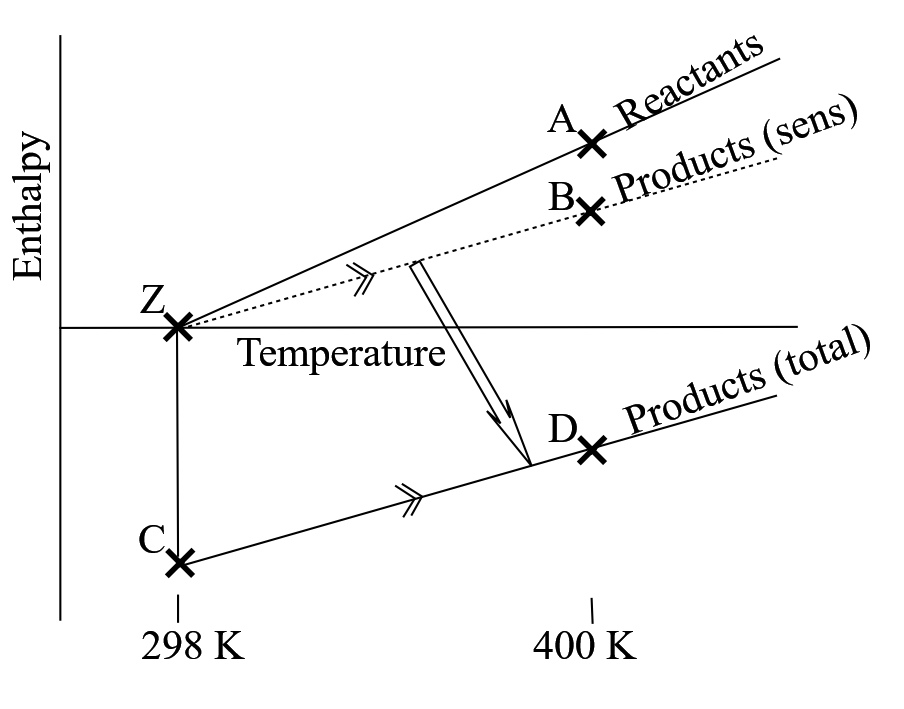
(I have assumed perfect gas behaviour and omitted superscript ø from enthalpy.) Figure 5 shows a diagramatic proof. Suppose the change in enthalpy from A to D is achieved via the ficticious path A-Z-C-D.
I choose location Z as the datum; it is part of the transition from hot reactants (A) to cool reactants (Z) to cool products (C). Line CD is sub-path from cool products to hot products. Line BZ is translated vertically to become CD so that path A-Z-C-D is contiguous. Distance DB matches \(ZC \equiv \Delta \widetilde{h}_o \), the standard enthalpy of combustion. Distance BA matches the difference in sensible enthalpy at temperature T.
Figure 5 Change in enthalpy at artibrary temperature. pø and in the pure condition. The curve for product sensible enthalpy is translated from ZB to CD \(BD = ZC \equiv \Delta \widetilde{h}_o^{\text{ø}} \) and
\( AB \equiv H^{sens}_P-H^{sens}_R = \sum \Delta n_i \times \widetilde{h}_{sensible,i,T} \)
Example 4B0.040: For the formation of nitric oxide (below) find the enthalpy of reaction and Gibbs function of reaction at (1)
standard pressure and temperature (2) standard pressure but T=2000 K.
½ N2 + ½ O2 ⇌ NO*
Solution: Sensible enthalpies are tabulated below. From the same source we obtain the enthalpy of combustion.
... the division of a molecule into smaller, simpler molecules, atoms, or ions, by a chemically reversible reaction.
I shall write the oxidation of carbon monoxide as a reversible reaction.
CO + ½ O2 ⇌ CO2
At high temperatures dissociation reverses the above reaction and produces small amounts of CO and \(O_2\). For example, at 1200K then 17 parts per billion of CO2 at 1 bar will dissociate. An
equilibrium constant \(K_T^{\text{ø}}\) relates the partial pressures to each other,
$$ K_T^{\text{ø}} = \frac{p_{co2}/p^{\text{ø}}}{ (p_{co}/p^{\text{ø}}) \times (p_{o2}/p^{\text{ø}})^{1/2}} $$
and the equilibrium constant \(K_T^{\text{ø}}\) follows from
$$ K_T^{\text{ø}} = exp ( -\frac{\Delta \widetilde{g}^ø_T}{\widetilde{R} T}) $$
where \( \Delta \widetilde{g}^ø_T \) is evaluated at a reference pressure \(p^{\text{ø}}\) and temperature T.
When evaluated for their partial pressures at
equilibrium, the Gibbs functions of the reactants and products are equal.
To obtain the above results Van't Hoff imagined that the Gibbs free energy was diverted to compress or expand the chemical species between their actual partial pressure and the reference pressure \( p^{\text{ø}} \).
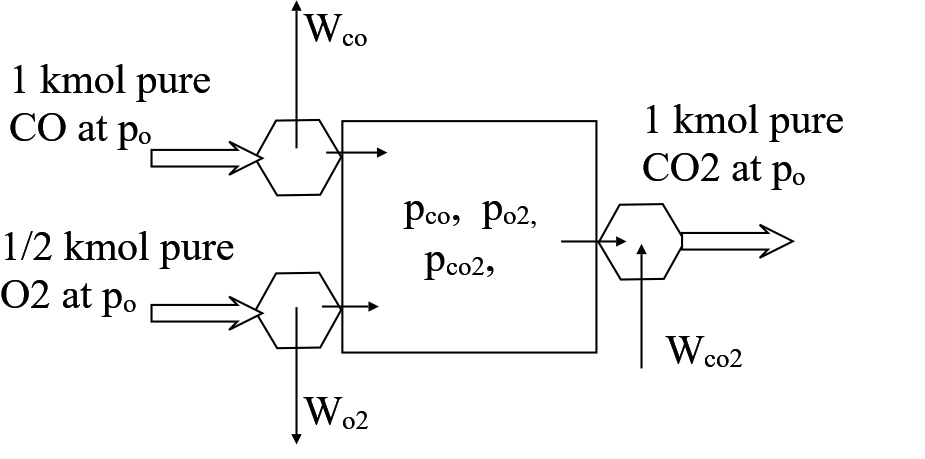
Figure 6 Van't Hoff box. Net work available from isothermal expansion and compression pure gas is equal to Gibbs function of reaction at standard pressure pø. (Work transfer is shown but corresponding heat transfer is omitted.) All gas is at the same temperature throughout. Gas enters/ leaves the box at pressure pø and in the pure condition.
Van't Hoff (Figure 6).
The box is fed with pure CO and pure O2, both at pø. Pure CO2
emerges from the box, also at pø. The reaction achieves its equilibrium inside the box so that so that all chemical species are present and exert partial pressures, pco, pco2 and po2. The hypothetical expanders and compressor at the edges of the box have a special reversible behaviour - only a pure gas will
pass through them. Thus CO is expanded isothermally from pø to pco, likewise the O2
from pø to po2 and CO2 is compressed isothermally from pco2 to pø. The net work of expansion is equated to the Gibbs function of reaction at temperature T.
We can further show that when taken at their equilibrium partial pressures the Gibbs functions of the reactant and product are equal. Expand the left hand side of Equation (A)
$$ \widetilde{g}_{T,co2}^ø - \widetilde{g}_{T,co}^ø - (1/2) \widetilde{g}_{T,o2}^ø
= - \widetilde{R} T ln (\frac{p_{co2}}{p^ø} ) + \widetilde{R} T ln( \frac{p_{co}}{p^ø}) + \frac{1}{2} \widetilde{R} T ln (\frac{p_{o2}}{p^ø}) ) $$
Rearrange so as to (1) group chemical species, and (2) put reactants on the LHS and product on the RHS.
$$
[\widetilde{g}_{T,co}^ø + \widetilde{R} T ln (\frac{p_{co}}{p^ø} )] +
(1/2)[ \widetilde{g}_{T,o2}^ø + (\widetilde{R} T ln (\frac{p_{o2}}{p^ø})]
= [\widetilde{g}_{T,co2}^ø + \widetilde{R} T ln (\frac{p_{co2}}{p^ø} )] $$
Each term in square brackets [] represents a Gibbs function at the prevailing partial pressure.
For example the first term contains the Gibbs function of carbon monoxide at
standard pressure ø (1 bar) plus the reversible isothermal work required to compress
the CO to a pressure \(p_{co}\), that is \( \widetilde{g}_{T,co}^ø + \widetilde{R} T ln (\frac{p_{co}}{p^ø} ) = \widetilde{g}_{T,co}^{p_{co}} \). Consider all three chemical species,
Under equilibrium conditions, the respective Gibbs functions of the reactants and products are equal.
For many energy balances the small concentrations of reactants can be treated as trivial; however they have clear implications for public health. A precise estimate of partial pressure lies well beyond the scope of these notes; one would have to account for the coupled flow patterns, multiple chemical reactions, the mass transfer and chemical kinetics of flame fronts, and changes to gas composition in the exhaust system. Realistic models need to be embedded in Computational Fluid Dynamics (CFD) codes, demaning large computational resources. Such models should be used in conjunction with expensive experimental test facilities.
Example 4B0.050:
Consider a fuel-cell operating at standard state and an internal combustion engine operating at
a maximum temperature of 2000K. Comment of the formation of the free radical nitric oxide according to
½ O2 + ½ N2 ⇌ NO*
Assume that for both cases total pressure is 1.3 bar and representative partial pressures are 0.1 bar for oxygen and 0.4 bar for nitrogen.
Solution: Start with \(T=298K\)
Use Gibbs function of reaction from previous Example 4B0.050 to obtain the
equilibrium constant,
This corresponds to a volume fraction of 3070 ppm. HOWEVER, nitric oxide is a free radical. It will rapidly react with nitrogen, oxygen and other radicals to form NOx. It is possible that these reactions are so fast the the level of NO* itself is below its nominal
equilibrium value, but nonetheless the high value here must be of concern, and justifies more detailed (and expensive) assessment.
Appendix
Preamble
The main text covers material that will be found in Keenan, or Rogers and Mayhew etc and
is adequate for the professional purposes of most Mechanical, Civil, and Aeronautical
Engineers. This appendix touches on material that might crop up in conversation during
an engineer's career, or might be used in specialist publications. Enthalpy of formation is specialist high school material, whereas
chemical potential is in the undergraduate Chemistry and Chemical Engineering syllabus.
Enthalpies of Formation
For most problems in the energy sector, it is usually sufficient to
have access to enthalpies of reaction . More widely, such reaction enthalpies can be
found, when required, from their enthalpies of formation.
\begin{align*}
\Delta \widetilde{h}^{\text{ø}}_o &= \sum \Delta n_i \times \Delta \widetilde{h}^{\text{ø}}_{f,o,i} \qquad 1kmol \; fuel \; at \; T_o, \; p^{\text{ø}} \; throughout \\
\Delta \widetilde{g}^{\text{ø}}_o &= \Delta \widetilde{h}^{\text{ø}}_o - T_o \sum \Delta n_i \times s ^{\text{ø}}_{o,i} \qquad 1kmol \; fuel \; at \; T_o, \; p^{\text{ø}} \; throughout \\
\end{align*}
The term \( \Delta \widetilde{h}^{\text{ø}}_{f,o,i} \) refers to the enthalpy of formation (f) at standard temperature (o) and pressure (ø) of species i. This is the enthalpy change when the chemical species is formed from elements in their natural state (O2, N2, H2, C); at the standard state the formation energies of said elements should be equal to zero (i.e. a change from N2 to N2 has no effect).
Example, consider the incomplete combustion of methane at the standard state.
Then the heat of reaction is,
$$ \Delta \widetilde{h}^ø_o = -110.53+2\times(-241.83) -1\times(-74.6) -1.5 \times 0 = -519.6 kJ/kmol $$
Chemical Potential
The chemical potential is the the differential molar free energy added by one particular chemical species, with temperature, pressure and amounts of all other species fixed.
Some academic papers tend to treat it synonymously with Gibbs function. Chemical potential is important when we consider (1) the dissociation of non-ideal gases (2) equilibrium between different phases (e.g. vapour-liquid).
Changes at constant temperature and pressure
At chemical equilibrium in a non-ideal gas, changes in chemical potential sum to zero.
Refer back to the Van't Hoff box (Figure 6). Let the contents be at constant pressure and constant temperature. A net addition to the box for each species of \(n_i\) kmol, with \( i =1,2,3\) here, produces a change in Gibbs function,
$$ \Delta G = n_1 \widetilde{g}_1^{p1} + n_2 \widetilde{g}_2^{p2} + n_3 \widetilde{g}_3^{p3} \qquad constant \; temperature \; and \; total \; pressure$$
This however assumes an ideal gas. For example, \( \widetilde{g}_1^{p1} \) is evaluated at its partial pressure and temperature following integration of the ideal gas law; it is also independent of the concentrations of species 2 or 3. In the non-ideal case the species Gibbs-functions are replaced with chemical potential. The possible interdependencies of chemical potentials and concentrations force the use of differential quantities,
$$ d G = dn_1 \mu_1 + dn_2 \mu_2 + dn_3 \mu_3 \qquad constant \; temperature \; and \; total \; pressure\ (A1)$$
At chemical equilibrium \(dG = 0 \).
Gibbs function with no chemical effects
Now consider variations in T,p. Let us "block" the compressors and expanders so that no material is fed to or taken from the Van't Hoff box (Figure 6). Also ignore any chemical effect therein, so that any gas mixture behaves in the same way as a pure gas. We know already that the Gibbs function is,
$$ G = H - TS = U + pV - TS \qquad no \; chemical \; effect \;(A2) $$
where TdS is due to reversible heat transfer and pdV is due to reversible work. Then substitution of Equation A2 into Equation A3 yields another expression for change in Gibbs function,
$$dG = -SdT + Vdp \qquad (A4) \qquad no \; chemical \; effect \;(A4)$$
That is, G is brought about by changes in pressure or temperature so long as the system is closed.
Equation A4 is nice, because an energy potential (G) depends on easily measured variables - temperature and pressure.
Gibbs function in general
Two different sets of processes require two separate equations for dG - Equations (A1) and (A4) above. The merger of these equations offers a wider generality.
$$ dG = -SdT + V dp + \mu_{1} d n_{1} + \mu_{2} d n_{2} + \mu{3} d n{3} $$
Returning to the case of constant temperature and pressure, a more pleasing defininition of chemical potential \( \mu \) arises. it becomes the differential free energy of one particular chemical species, with temperature, pressure and amounts of all other species fixed. (It is also referred to as a 'partial molar' property - the change in an extensive property in a large reservoir of gas mixture following the addition of 1 kmol at the same temperature and pressure.) Recall that Gibbs Free Energy is the potential of a chemical mixture to do work at constant temperature and pressure.
(the subscript referring to temperature is omitted). The simple mathematical form has
appealed to scientists, who (put very bluntly) have forced concepts to fit the equation.
Now at high pressures gases are not ideal and \( \int V dp \) does not yield the required logarithm.
Recall that at high pressure a compressibility factor applies, hence,
$$ pV = Z n \widetilde{R}T $$
A special term for gases is fugacity, f, computed from function V(p) in such a way that,
And often the reference condition is at sufficiently low pressure for \( f^ø \rightarrow p^ø \). If we take this to be so then,
$$ \Delta \widetilde{g}^ø = \sum n_i \widetilde{g}_i^ø \rightarrow \sum n_i \mu_i^ø \qquad at \; reference \; pressure $$
and, taking an earlier example
$$ K_T^{\text{ø}} = exp(-\frac{\Delta \widetilde{g}^ø }{RT}) =
\frac{f_{co2}/p^ø}{ (f_{co}/p^ø ) \times (f_{O2}/p^{\text{ø}})^{1/2}}
$$
Fugacity is described elsewhere as
"the effective partial pressure" . In other words, in some aspects of Chemical Engineering
the equilibrium constant would be related to fugacities.
(See this link.)
Fugacity is a special instance of "activity", which is important to chemical equilibria in the
liquid-phase. The importance of fuel cells and electrolyic cells to sustainability will drive Mechanical Engineers to take an interest in electrochemistry and liquid electrolytes. Again the logarithmic term is "fudged" by replacing the pressure ratio with an activity . We need to specify new aspects of datum conditions such as the molarity of the datum solution. I shall use a new symbol * for this purpose.
where \(a_i\) is termed the activity and \( \gamma_i \) is termed the activity coefficient. For liquids it may well be computed from molar concentrations (molarities), rather than partial pressures. In electrochemistry activity tells us the ionic strength a solution.